What is bioinformatics?
▪Can be defined as the body of tools, algorithms needed to
handle large and complex biological information.
▪ Bioinformatics is a scientific discipline created from the
interaction of biology and computer science.
▪ The technology that uses computers for the storage, retrieval,
manipulations and distribution of information related to bio-
macromolecules i.e., RNA/DNA, proteins.
▪ An algorithm is a finite sequence of rigorous instructions,
typically used to solve a class of specific problems or to
perform a computation
3.
Biologists
collect molecular data:
DNA& Protein sequences,
gene expression, etc.
Computer scientists
(+Mathematicians, Statisticians, etc.)
Develop tools, softwares, algorithms
to store and analyze the data.
Bioinformaticians
Study biological questions by
analyzing molecular data
The field of science in which biology, computer science and information
technology merge into a single discipline
3
4.
4
Bioinformatics in basicresearch
Large amount of genomic data
Storage/management/analysis sequeces are sorted &
assembled softwares assembled data makes sense
Annotation software search for functional signals in the
genomes to infer coding genes in the sequence and other
type of functional non-coding sequences.
Genome annotation is the process of identifying functional elements along the sequence
of a genome, thus giving meaning to it
5.
5
Comparative and evolutionarygenomics
▪ Genomes comparison of close/distant
species to unravel the evolutionary
processes that occur in the genome.
▪ It possible to be known from the
conserved sequences between species,
which are the genome functional parts.
6.
6
Functional genomics andother omics
▪ Functional genomics is the comprehensive analysis of
function, expression and interaction of all genes in an
organism.
▪ The term genome (gene + ome, where ome is
understood "all genes"), other “omics” terms have been
used to describe the study of other global data sets.
▪ The transcriptome (the sequences and expression
patterns of all transcripts),
▪ The proteome (the sequences and expression patterns of
all proteins), the interactome (the complete set of
physical interactions between proteins, DNA sequences
and RNA), The epigenome (the complete set of epigenetic
modifications on the genetic material of a cell), are some
examples.
7.
7
Genome Wide Associationanalysis
▪ To find out what genetic variants make us different each
other within our specie it is necessary to study the
genomes of many individuals.
▪ The HapMap project to characterize genetic variation
patterns in different ethnic groups of the human
species, as a preliminary step to take on genome-wide
studies, associated genetic variants with different
aspects on the phenotype, especially those that confer
susceptibility to disease.
▪ The comprehensive catalog of genetic variants affecting
human phenotype, with their enormous implications
arising for prevention, diagnosis and personalized
treatment of diseases.
8.
8
Biomedicine
▪ The humangenome has profound effects on clinical
medicine as every disease has a genetic component
▪ We can search for the genes directly associated with
different diseases and begin to understand the
molecular basis of these diseases more clearly
▪ The molecular mechanisms of disease will enable better
treatments, cures and even preventative tests to be
developed.
9.
9
Drug discovery
▪ Usingcomputational tools to identify
and validate new drug targets will help in
more specific medicines.
▪ These highly specific drugs will have
fewer side effects than many of today's
medicines.
10.
10
Personalized medicine
▪ Pharmacogenomicsstudy will help to find how
an individual's genetic inheritance affects the
body's response to drugs
▪ Doctors will be able to analyze a patient's genetic
profile and prescribe the best available drug
therapy and dosage from the beginning
11.
11
Agriculture
▪ Bioinformatics toolscan be used to search for the genes
within the plant genomes and to elucidate their functions
▪ This specific genetic knowledge could then be used to
produce stronger, more drought, disease and insect
resistant crops and improve the quality of livestock
making them healthier, more disease resistant and more
productive.
12.
Database/Biological Database
Databaseare convenient system to properly store, search and retrieve any type of
data.
A database helps to easily handle and share large amount of data and supports
large scale analysis by easy access and data updating.
Biological databases are libraries of life sciences information, collected from
scientific experiments, published literature, high throughput experiment technology
and computational analysis.
They contain information from genomics, proteomics, microarray gene expression.
Information contained in biological databases includes gene function, structure,
localization(both cellular and chromosomal), biological sequences and structures.
13.
Primary databases
Thesesare the primary sources of data used to store nucleic acid, protein
sequences and structural information of biological macromolecules, e.g.,
NCBI (The National Centre for Biotechnology Information)
Gene Bank
DDBJ (DNA data bank of Japan)
SWISS-PROT (Swiss-Prot )
PIR (Protein Information Resource)
PDB (Protein Data Bank)
This sequence collection of this database is due to the efforts of basic research
from academic industrial and sequencing lab
14.
Secondary Database
ASecondary database contain additional information derived from the
analysis of data available in primary sources.
Much of this information is obtained from scientific literature and entered by
database curators e.g.,
TrEMBL
Pfam
PROSITE
NCBI Structures
RefSeq
CATH
15.
Specialized Databases
Theyserve a specific research community/focus on a particular organism.
The sequences in these databases may overlap with a primary database, but
may also have new data submitted directly by authors e.g.,
HIV databases
Microarray gene expression database
TAIR (Arabidopsis information database)
16.
Interconnecting Prim &Sec Database
To upload info in sec. database prim. & Sec database needs
interconnection.
To complete a task information in a single database are insufficient so need
interconnection.
Entrees in both databases may be cross referenced to avoid searching in
multiple databases.
17.
Barriers/solutions to interconnecting
databases
Format incompatibility
Heterogeneous structures (flat file, relational & object oriented) of
databases limits communication between databases.
Common Object Request Broker Architecture (COBRA), which allows
database programs at different locations to communicate in a network
through an “interface broker” without having to understand each other’s
database structure.
eXtensible Markup Language (XML) can also help in bridging databases
Recently, a specialized protocol for bioinformatics data exchange has been
developed.
18.
Drawbacks/solution of Bio-databases
Chances of error
Redundancy
Gene annotations may be wrong/incomplete
Erroneous annotations of genes
NCBI has created nonredundant database called RefSeq
To overcome redundancy sequence-cluster databases such as UniGene created
19.
Database Retrieval
Retrievalof complex info req. Boolean operators i.e.,
AND. Use AND to narrow your search: all of your search terms will present in the
retrieved records. ...
OR. Use OR to broaden your search by connecting two or more synonyms.
NOT. Use NOT to exclude term(s) from your search results.
Parentheses ( ) to define a concept if multiple words and relationships are
involved, so that the computer knows which part of the search to execute first.
20.
Database retrieval
DatabasesRetrieval/Systems Brief Summary of Content URL
AceDB Genome database for Caenorhabditis elegans: www.acedb.org
DDBJ Prim nucleotide seq. database: www.ddbj.nig.ac.jp
EMBL Prim nucleotide seq. database: www.ebi.ac.uk/embl/index.html
Entrez NCBI portal for biodatabases: www.ncbi.nlm.nih.gov/gquery/gquery.fcgi
ExPASY Proteomics database: http://us.expasy.org/
GenBank Prim nucleotide seq. database www.ncbi.nlm.nih.gov/Genbank
OMIM Genetic informations of human diseases
www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM
21.
NCBI
One ofthe most useful and comprehensive sources of databases is the NCBI, part of
the National Library of Medicine, funded by USA.
NCBI provides interesting summaries, browsers for genome data, and search tools
Gateway to search txt based searches including genetic sequence information,
structural information, citations, abstracts, full papers, and taxonomic data.
Cross-referencing NCBI databases based on preexisting and logical relationships
between individual entries e.g., in a nucleotide sequence page, one may find cross-
referencing links to the translated protein sequence, genome mapping data, or to
the related PubMed literature information, and to protein structures if available.
22.
Limits,” whichhelps to restrict the search to a subset of a particular database
e.g., the field for author or publication date.
“Preview/Index,” which connects different searches with the Boolean perators
and uses a string of logically connected keywords to perform a new search.
“History” option provides a record of the previous searches.
“Clipboard” that stores search results for later viewing for a limited time.
23.
NCBI
ClinVar (www.ncbi.nlm.nih.gov/clinvar/)
▪Medical genetics resource that collects assertions of the relationships between
human sequence variations and phenotypes
▪ Submissions to ClinVar may specify the variation, the phenotype, the
interpretation of the medical importance of the variation, the date that
interpretation was last evaluated and the evidence supporting that
interpretation, along with information about the submitter.
▪ Each of the individual assertions submitted to ClinVar has a unique accession of
the format SCV000000000.0, and submissions that relate the same variant and
phenotype are collected in reference records with accessions
RCV000000000.0
24.
NCBI
MedGen (www.ncbi.nlm.nih.gov/medgen/)
MedGen organizes information about phenotypes around a stable identifier
assigned to terms used to name disorders and their clinical features.
MedGen uses a combination of automatic processing and curation to aggregate
these data, and presents the results as a text report with several section, may
include, depending on the available data, descriptions of the disease and its
clinical features along with collections of relevant professional guidelines, clinical
studies and systematic reviews
PubReader: NCBI’s reader-friendly display option for viewing full-text articles in
the PubMed Central (PMC) database
Medical Subject Headings (MeSH) database includes information about the NLM
controlled vocabulary thesaurus used for indexing PubMed citations
25.
NCBI
PopSet isa collection of related sequences and alignments derived from population,
phylogenetic, mutation and ecosystem studies that have been submitted to GenBank
SRA (Sequence Read Archive) is a repository for raw sequence reads and alignments
generated by the latest generation of high throughput nucleic acid sequencer
Biosystems database collects together molecules represented in Gene, Protein and
PubChem that interact in a biological system such as a biochemical pathway or
disease
Gene Expression Omnibus (GEO) is a data repository and retrieval system for high-
throughput functional genomic data generated by microarray and next-generation
sequencing technologies
GEO Profiles, which contains quantitative gene expression measurements for one
gene across an experiment
GEO DataSets, contains entire experiments.
26.
NCBI
UniGene isa system for partitioning transcript sequences (including ESTs) from
GenBank into a nonredundant set of clusters
HomoloGene is a system that automatically detects homologs, including paralogs
and orthologs, among the genes of 21 completely sequenced eukaryotic
genomes.
The Probe database is a registry of nucleic acid reagents designed for use in a
wide variety of biomedical research applications including genotyping, SNP
discovery, gene expression, gene silencing and gene mapping
The Database of Genotypes and Phenotypes (dbGaP) archives, distributes and
supports submission of data that correlate genomic characteristics with
observable traits
Orthologs are homologs in different species that catalyze the same reaction,
and paralogs are defined as homologs in the same species that do not catalyze
the same reaction
27.
NCBI
The Databaseof Genomic Structural Variation (dbVar) is an archive of large-
scale genomic variants (generally >50 bp) such as insertions, deletions,
translocations and inversions
The Database of Short Genetic Variations (dbSNP) is a repository of all types
of short genetic variations <50 bp in length, and so is a complement to dbVar
28.
GenBank Sequence Format
Header section describes the origin of the sequence, identification of the
organism, and unique identifiers associated with the record.
Locus, which contains a unique database identifier for a sequence location in the
database (not a chromosome locus). The identifier is followed by sequence length
and molecule type (e.g., DNA or RNA).
DEFINITION,” provides the summary information for the sequence record including
the name of the sequence, the name and taxonomy of the source organism if
known, and whether the sequence is complete or partial.
ACCESSION NUMBER a unique number assigned to a piece of DNA when it was
first submitted to GenBank and is permanently associated with that sequence.
29.
Protein Domains andMacromolecular
Structures
The resources developed by the Protein Classification and Structure
Group of the Information Engineering Branch (IEB) are freely available
to the public and focus on two primary areas.
Conserved domains: Conserved domains are functional units within a
protein that act as building blocks in molecular evolution and
recombine in various arrangements to make proteins with different
functions.
The Conserved Domain Database (CDD) brings together several
collections of multiple sequence alignments representing conserved
domains, in addition to NCBI-curated domains that use 3D-structure
information explicitly to define domain boundaries and provide insights
into sequence/structure/function relationships.
31
Primary Structure -Amino Acids
AA sequence in a
protein
The AA sequence
“exclusively”
determines the 3D
structure of a protein
20 amino acids –
modifications do occur
post transnationally
32.
32
Amino Acids Continued…
Chirality – amino acids are
enatiomorphs, that is mirror
images exist – only the L(S) form
is found in naturally forming
proteins. Some enzymes can
produce D(R) amino acids
Data structure for this
information – annotation and a
validation procedure should be
included
Primary Structure
33.
Amino acids
Polar,uncharged amino acids
Contain R-groups that can form hydrogen bonds with water
Includes amino acids with alcohols in R-groups (Ser, Thr, Tyr)
Amide groups: Asn and Gln
Usually more soluble in water
◼ Exception is Tyr (most insoluble at 0.453 g/L at 25 C)
Sulfhydryl group: Cys
◼ Cys can form a disulfide bond (2 cysteines can make one
cystine)
34.
Amino acids
Acidicamino acids
Amino acids in which R-group contains a carboxyl group
Asp and Glu
Have a net negative charge at pH 7 (negatively
charged pH > 3)
Negative charges play important roles
◼Metal-binding sites
◼Carboxyl groups may act as nucleophiles in
enzymatic interactions
◼Electrostatic bonding interactions
35.
Amino acids
Basicamino acids
Amino acids in which R-group have net positive charges
at pH 7
His, Lys, and Arg
Lys and Arg are fully protonated at pH 7
◼Participate in electrostatic interactions
His plays important roles as a proton donor or acceptor
in many enzymes.
His containing peptides are important biological buffers
36.
Nonstandard amino acids
20 common amino acids programmed by genetic
code
Nature often needs more variation
Nonstandard amino acids are usually the result of
modification of a standard amino acid after a
polypeptide has been synthesized.
Nonstandard amino acids play a variety of roles:
structural, antibiotics, signals, hormones,
neurotransmitters, intermediates in metabolic cycles,
etc.
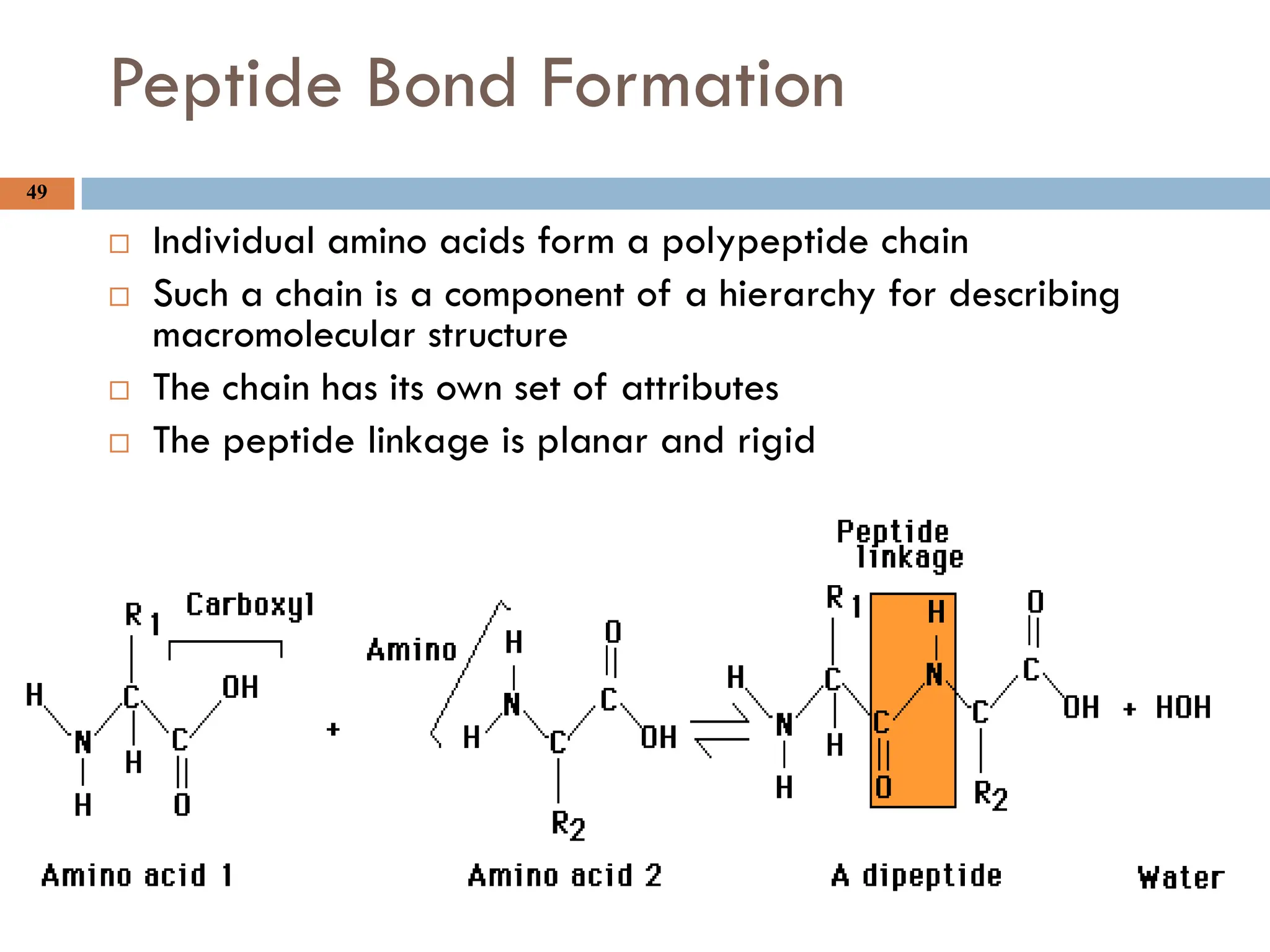
Peptide bonds
Proteins aresometimes called polypeptides since they contain many peptide bonds
H
C
R1
H3N
+
C
O
OH N
H
H
C
R2
O-
C
O
H
+
H
C N
R1
H3N
+
C
O
H H
C
R2
O-
C
O
+ H2O
42.
Structural character ofamide groups
Understanding the chemical character of the amide is important since the
peptide bond is an amide bond.
These characteristics are true for the amide containing amino acids as well
(Asn, Gln)
Amides will not ionize:
O O
R C NH2 R C NH2
43.
All aminoacids are optically active (exception Gly).
Optically active molecules have asymmetry; not superimposable (mirror images)
Central atoms are chiral centers or asymmetric centers.
Enantiomers -molecules that are nonsuperimposable mirror images
Amino acids are optically active
44.
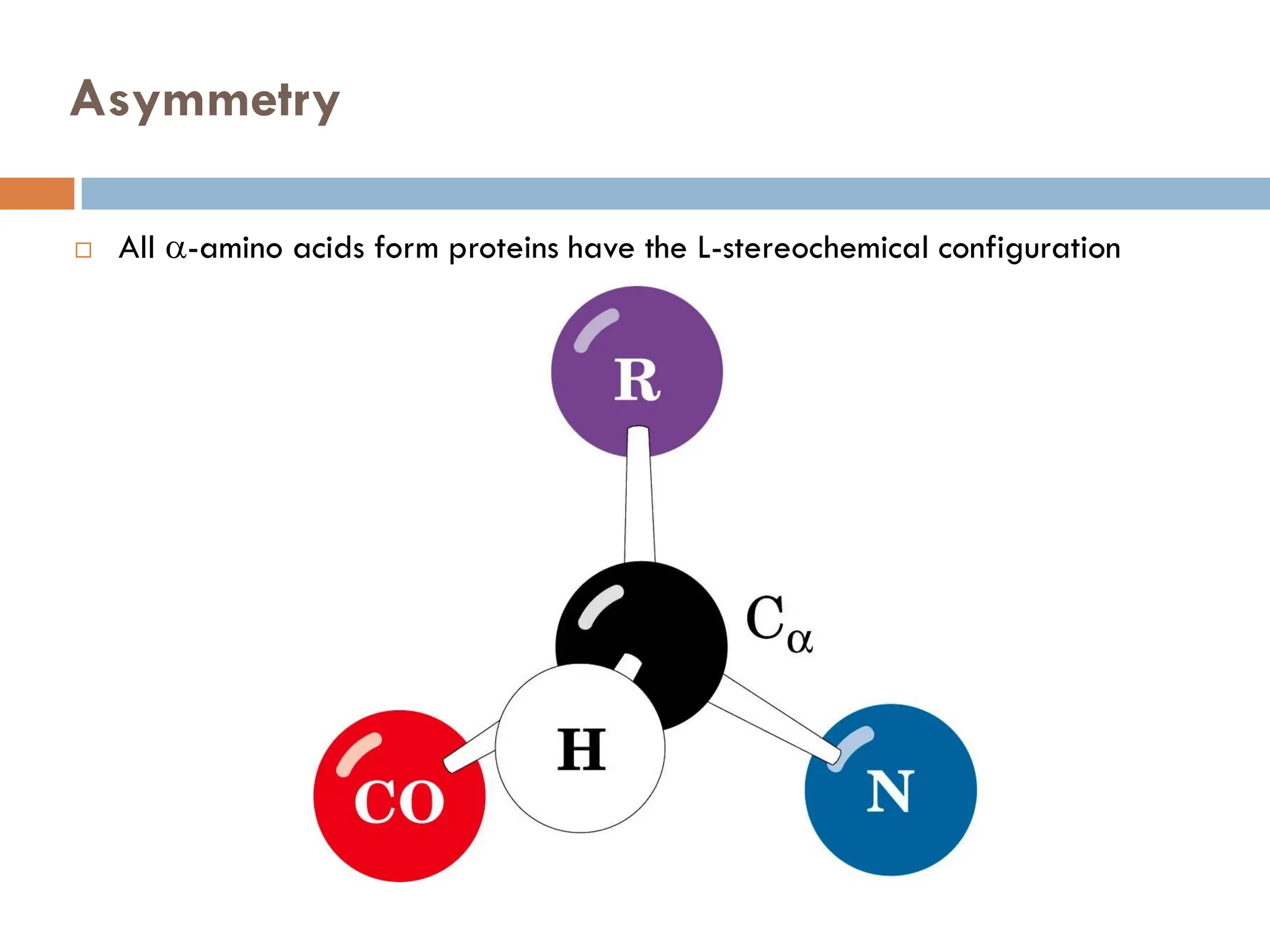
Asymmetry
Molecules areclassified as Dextrorotatory (right handed), D or Levorotatory (left
handed) L depending on whether they rotate the plane of plane-polarized light
clockwise or counterclockwise determined by a polarimeter
Diastereomers, Enantiomers &Meso
Diastereomers: Non-superimposable, Non-mirror images
Enantiomers: Non-superimposable, Mirror images
Meso: A molecule that is superimposable on its mirror image is optically inactive
49
Peptide Bond Formation
Individual amino acids form a polypeptide chain
Such a chain is a component of a hierarchy for describing
macromolecular structure
The chain has its own set of attributes
The peptide linkage is planar and rigid
50.
50
Geometry of theChain
A dihedral angle is the angle
between two planes defined by 4
atoms – 123 make one plane; 234
the other
Omega is the rotation around the
peptide bond Cn – Nn+1 – it is
planar and is 180 under ideal
conditions
Phi is the angle around N – C alpha
Psi is the angle around C-alpha-C’
The values of phi and psi are
constrained to certain values based
on steric clashes of the R group. Thus
these values show characteristic
patterns as defined by the
Ramachandran plot
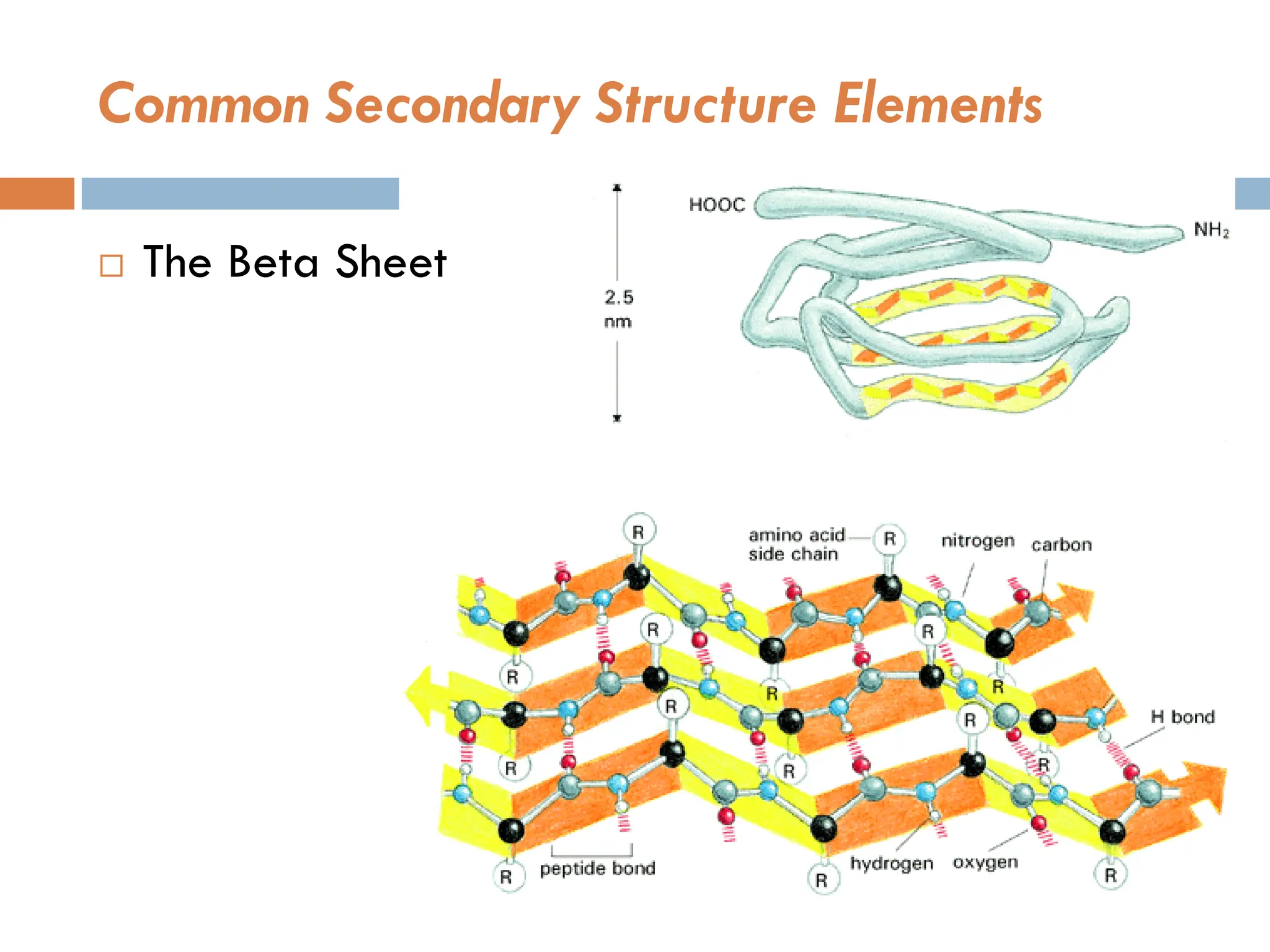
Secondary Structure
51.
Dihedral Angles
Theangle between two
intersecting planes.
In chemistry it is
the angle between planes
through two sets of three atoms,
having two atoms in common.
In solid geometry it is defined
as the union of a line and two
half-planes that have this line as
a common edge.
52.
52
Properties of alphahelix
Linus Pauling predicted the existence of α-
helices
There are 3.6 residues per turn means one
residue for every 100 degrees of rotation
Each A. A residue is at a distance of 1.5 Å
There is a H-bond between C=O of ith residue
& -NH of (i+4)th residue
H-bond between C=O of ith residue & -NH of
(i+4)th residue
First -NH and last C=O groups at the ends of
helices do not participate in H-bond
Ends of helices are polar, and almost always at
surfaces of proteins
Always right- handed
Secondary Structure
Since the dipolemoment of a peptide bond is 3.5 Debye units, the alpha
helix has a net macrodipole of:
n X 3.5 Debye units (where n= number of residues)
This is equivalent to 0.5 – 0.7 unit charge at the end of the helix.
Basis for the helical dipole
In an alpha helix all of the peptide
dipoles are oriented along the
same direction.
Consequently, the alpha helix has
a net dipole moment.
The amino terminus of an alpha helix is positive and the
carboxy terminus is negative.
THE RAMACHANDRAN PLOT
N-Calpha and Calpha-C bonds relatively are free to rotate
Ramachandran used computer models of small polypeptides to
systematically vary phi and psi with the objective of finding stable
conformations
Phi and Psi angles which cause spheres to collide correspond to sterically
disallowed conformations of the polypeptide backbone
58.
White regions: sterically
disallowedfor all amino
acids except glycine
Red region: no steric clashes allowed
regions namely the alpha-helical
and beta-sheet conformations.
yellow region: Allowed regions if slightly
shorter van der Waals radi are used
61.
BLAST (Basic LocalAlignment Search Tool)
Take a sequence and search for related sequence in
large databases
Find appropriate BLAST program → Entry Query
Sequence → Select database → Run BLAST →
Analyze output → Interpret E-value
62.
Multiple Sequence Alignment
Multiple Sequence Alignment (MSA) is generally the alignment of
three or more biological sequences (protein or nucleic acid) of similar
length.
Show homolog, evolutionary relationships
63.
ClustalW usedfor aligning multiple nucleotide or protein sequences
in an efficient manner. It align the most similar sequences first and
work their way down to the least similar sequences until a global
alignment is created
64.
Multiple Sequence Alignment(ClustalW)
Alignment of more than two sequences
From where we will take sequences?
Where to paste sequences in input page?
What parameters where to adjust?
Application
• Can make phylogenetic tree.
• Primer designing
65.
Phylogenetic Tree (Dnd)and anatomy
A diagram that represents
evolutionary relationships among
organisms. Phylogenetic trees are
hypotheses, not definitive facts.
The species or groups of interest
are found at the tips of lines
referred to as the tree's branches.
The branches pattern represent how
the species in the tree evolved from
a series of common ancestors.
66.
Dendogram: Tree
diagram
Cladograms:
Phylogenies that depict
only branching order
Types of cladogram
Branching order is
important not length in
cladogram
67.
Cladogram and phylogram
a is cladogram with no effect of line length
Phylograms typically include a scale bar
to indicate how much change is reflected
in the lengths of the branches
b is phylogram,” in which branch length is
proportional to some measure of
divergence e.g., V more diverged than U
c, the terminal nodes are aligned with
each other and the internal branch lengths
are scaled to show the degree of
divergence among sister groups rather
than among individual species
68.
Primer
Short singlenucleic acid sequence that provides a starting point for DNA
synthesis.
The leading strand is synthesized in continuous fashion, requiring only an initial
RNA primer to begin synthesis while in lagging strand, the template DNA runs in
the 5′→3′ direction.
In lagging strand DNA is synthesized ‘backward’ in short fragments moving
away from the replication fork, known as Okazaki fragments.
In lagging strand the repeated starting and stopping synthesis of DNA, requires
multiple RNA primers.
Synthetic primers are chemically synthesized oligonucleotides, usually of DNA,
which can be customized to anneal to a specific site on the template DNA.
The primer spontaneously hybridizes with the template through Watson-Crick
base pairing before being extended by DNA polymerase
69.
PCR primer design
Primer design is aimed at obtaining a balance between specificity
and efficiency of amplification.
❖ Specificity is the ability of a primer to correctly identify and pair.
❖ Primers with poor specificity tend to produce PCR products with
extra unrelated and undesirable amplicons.
❖ Efficiency is defined as how close a primer pair is able to amplify a
product to the theoretical optimum of a twofold increase of product
for each PCR cycle.
70.
Primer Length
Thespecificity and efficiency depends on primer length and annealing
temperature of the PCR reaction i.e., 18 and 24 bases primer
Specificity: Increased primer length increases specificity but decreasing efficiency
Tm (defined as the dissociation temperature of the primer/template duplex),
Optimal range of temperature is from 54-65 oC
Short oligonucleotides of 15 bases or less are useful only for a limited amount of
PCR protocols and for mapping simple genome. In general, it is best to build in a
margin of specificity for safety.
For each additional nucleotide, a primer becomes four times more specific; thus,
the minimum primer length used in most applications is 18 nucleotides.
71.
Base Composition andTm
Usually, average (G+C) content around 40-60% will give us the right
melting/annealing temperature (Tm) values in the range of 40-60 oC and will give
appropriate hybridization stability.
Within a primer pair, the GC content and Tm should be well matched. Poorly
matched primer pairs can be less efficient and specific because loss of specificity
arises with a lower Tm and the primer with the higher Tm has a greater chance of
mispriming under these conditions.
Matching of GC content and Tm is critical when selecting a new pair of primers
from a list of already synthesized oligonucleotides within a sequence of interest
for a new application.
72.
The Terminal Nucleotidein Primer
3'-terminal position in the primer is essential for controlling mispriming.
74.
Automated Primer Design:Primer 3
Input protocol
Select gene---->Copy gene--->Pasted in box--->
--->set the parameters---->pick primer
75.
Expasy
ExPASy isthe Swiss Institute of Bioinformatics (SIB)
Bioinformatics Resource Portal which provides access to
scientific databases and software tools (i.e., resources) in
different areas of life sciences including proteomics, genomics,
phylogeny, systems biology, population genetics, transcriptomics
etc.
❖ . (http://www.expasy.org/tools/)
76.
Protein Identification andanalysis on Expasy
Protein identification and analysis software performs a
central role in the investigation of proteins from two-
dimensional (2-D) gels and mass spectrometry
For protein identification, the user matches certain
empirically acquired information against a protein
database to define a protein as already known or as novel
For protein analysis, information in protein databases can
be used to predict certain properties about a protein, which
can be useful for its empirical investigation.
Analysis tools include Compute pI/Mw, predicting protein
isoelectric point (pI)/Mw ProtParam, to calculate various
physicochemical parameters
77.
UniProt
UniProt isa freely accessible database of protein sequence and functional
information.
UniProt is the Universal Protein resource, a central repository of protein data
created by combining the Swiss-Prot, TrEMBL and PIR-PSD databases.
It contains a large amount of information about the biological function of
proteins derived from the research literature.
In 2002 a merge and collaboration of three databases;
European Bioinformatics Institute (EBI), Swiss Institute of Bioinformatics (SIB), and
Protein Information Resource (PIR)
❑ Recently EBI and SIB together produced the Swiss-Prot and TrEMBL databases
❑ PIR produced the Protein Sequence Database (PIR-PSD)
78.
Uniprot Databases
TheUniProt Knowledgebase (UniProtKB) is the central access point for extensive
curated protein information, including function, classification, and crossreference.
UniProtKB is divided into groups
❖ UniProtKB/Swiss-Prot which is manually curated
❖ UniProtKB/TrEMBL which is automatically maintained
❑ UniProt Archive (UniParc) is a comprehensive and non-redundant database which
contains all the protein sequences from the main, publicly available protein
sequence databases.
❖ UniParc contains only protein sequences, with no annotation.
❑ UniRef: consist of three databases of clustered sets
❖ UniRef100Combines identical sequences and sequence fragments (from any
organism) into a single UniRef entry.
❖ UniRef90: 90% identity
❖ UniRef50: 50% identity
79.
I-Tasser
I-TASSER (IterativeThreading ASSEmbly Refinement) is a hierarchical
approach to protein structure prediction and structure-based function
annotation.
Protein Structure Prediction
DO prediction through: Sequence similarity---Structure matching---Function
80.
Flow chart
Genefrom NCBI----Translate in Expasy (select
longest sequence----Copy and paste in I-Tasser
Note
Enter Email ID after clicking email ID can be
registered
Protein Threading
Proteinthreading or fold recognition, is a method of protein modeling
which is used to model those proteins which have the same fold as
proteins of known structures, but do not have homologous proteins with
known structure.
Threading works by using statistical knowledge of the relationship
between the structures deposited in the PDB and the sequence of the
protein which one wishes to model.
Generalization of homology modeling
Homology modeling: align sequence to sequence
Threading: align sequence to structure (templates)
❖ Basis of the idea of threading
❑ Limited number of basic folds found in nature
Most of the proteins has similar folds.
88.
The basicidea of protein threading is to place (align or thread) the amino acids
of a query protein sequence, following their sequential order and allowing gaps,
into structural positions of a template structure in an optimal way measured by
fitness scores.
This procedure will be repeated against a collection of previously solved protein
structures for a given query protein.
These sequence structure alignments, i.e., the query sequence against different
template structures, will be assessed using statistical or energetic measures for the
overall likelihood of the query protein adopting each of the structural folds.
The "best" sequence-structure alignment provides a prediction of the backbone
atoms of the query protein, based on their placements in the template structure.
89.
Advantage
Protein threadingis being widely used in molecular biology and biochemistry
labs, often for initial studies of target proteins, as it may quickly provide structural
and functional information, which could be used to guide further experimental
design and investigation.
90.
Challenges
(a) howto effectively and accurately measure the
fitness of a sequence placed in a template structure
(b) how to accurately and efficiently find the best
alignment between a query sequence and a template
structure based on a given set of fitness measures
(c) how to assess which sequence-structure alignment
among the ones against different template structures
represents a correct fold recognition and an accurate
(backbone) structure prediction, and
(d) how to identify which parts of a predicted
structure are accurate and which parts are not.
91.
Homology modeling withSWISS-MODEL
Homology modeling allows to build the structure of a protein when only its
amino acid sequence and the complete atomic structure of at least one other
reference protein is known.
Homology modelling methods make use of experimental protein structures
("templates") to build models for evolutionary related proteins ("targets")
The reference protein must be structurally homologous to the model protein
being build. Structural segments, which are thought to be conserved within the
family of homologous proteins are taken directly from the reference protein
Modeling of protein structures usually requires extensive expertise in structural
biology and the use of highly specialized computer programs for each of the
individual steps of the modeling process. The idea of an easy-to-use, automated
modeling facility with integrated expert knowledge was first implemented 12
years ago by Peitsch et al. and formed the starting point for the SWISS-MODEL
server.
92.
SWISS-MODEL isa structural bioinformatics web-server dedicated to homology
modeling of 3D protein structures.
3D protein structures provide valuable insights into the molecular basis of
protein function, allowing an effective design of experiments, such as site-
directed mutagenesis, studies of disease-related mutations or the structure
based design of specific inhibitors.
Automated homology modeling systems;
ModPipe (http://www.salilab.org)
CPHmodels (http://www.cbs.dtu.dk/services/CPHmodels/)
3D-JIGSAW (http://www.bmm.icnet.uk/~3djigsaw/)
ESyPred3D (http://www.fundp.ac.be/urbm/bioinfo/esypred/)
SDSC1 (http://cl.sdsc.edu/hm.html)].
93.
SWISS-MODEL MODES
TheSWISS-MODEL server is designed to work with a minimum of user input, i.e.
only the amino acid sequence of a target protein. As comparative modeling
projects can be of different complexity, additional user input may be necessary
for some modeling projects, e.g. to select a different template or adjust the
target-template alignment. The SWISS-MODEL server gives the user the choice
between three main interaction modes;
❖ Approach mode
❖ Alignment mode
❖ Project mode
94.
Approach mode
The‘first approach mode’ provides a simple interface and requires only an
amino acid sequence as input data. The server will automatically select suitable
templates.
The user can specify up to five template structures, either from the ExPDB library
or uploaded coordinate files. The automated modeling procedure will start if at
least one modeling template is available that has a sequence identity of more
than 25% with the submitted target sequence.
The model reliability decreases as the sequence identity decreases and that
target-template pairs sharing less than 50% sequence identity may often
require manual adjustment of the alignment.
95.
Alignment mode
Inthe ‘alignment mode’ the modeling procedure is
initiated by submitting a sequence alignment.
The user specifies which sequence in the given
alignment is the target sequence and which one
corresponds to a structurally known protein chain from
the ExPDB template library.
The server will build the model based on the given
alignment.
96.
Project mode
The‘project mode’ allows the user to submit a manually optimized
modeling request to the SWISS-MODEL server.
The starting point for this mode is a DeepView project file. It
contains the superposed template structures, and the alignment
between the target and the templates. This mode gives the user
control over a wide range of parameters, e.g. template selection or
gap placement in the alignment.
The project mode can also be used to iteratively improve the output
of the ‘first approach mode’.
97.
The Swiss modellingworkflow
Input data:
❖ The target protein can be provided as amino acid
sequence, either in FASTA, Clustal format or as a plain
text.
❖ A UniProtKB accession code can be specified.
❖ If the target protein is heteromeric, i.e. it consists of
different protein chains as subunits, amino acid
sequences or UniProtKB accession codes must be
specified for each subunit.
98.
Template search
InputData serve as a query to search for evolutionary
related protein structures against the SWISS-MODEL
template library SMTL.
❖ SWISS-MODEL performs this task by using two
database search methods:
▪ BLAST , which is fast and sufficiently accurate for
closely related templates, and
▪ HHblits, which adds sensitivity in case of remote
homology
99.
Template selection
❖ Aftertemplate search, templates are ranked according to expected
quality of the resulting models, as estimated by Global Model Quality
Estimate (GMQE) and Quaternary Structure Quality Estimate (QSQE).
❖ Top-ranked templates and alignments are compared to verify whether
they represent alternative conformational states or cover different
regions of the target protein.
❖ Multiple templates are selected automatically and different models are
built accordingly. To provide the user with the option to use alternative
templates than those selected automatically, all templates are shown in a
tabular form with a descriptive set of features.
❖ Interactive graphical views facilitate the analysis and comparison of
available templates in terms of their three-dimensional structures,
sequence similarity and quaternary structure features.
100.
Model building
❖ Foreach selected template, a 3D protein model is automatically
generated by first transferring conserved atom coordinates as
defined by the target template alignment.
❖ Residue coordinates corresponding to insertions/deletions in the
alignment are generated by loop modelling
❖ Full-atom protein model is obtained by constructing the non-
conserved amino acid side chains.
❖ SWISS-MODEL relies on the Open Structure computational
structural biology framework and the ProMod3 modelling engine to
perform this step.
101.
Model qualityestimation:
❖ To quantify modelling errors and give estimates on expected model accuracy,
SWISSMODEL relies on the QMEAN scoring function
❖ QMEAN uses statistical potentials of mean force to generate global and per
residue quality estimates.
❖ The local quality estimates are enhanced by pairwise distance constraints that
represent ensemble information from all template structures found.
102.
Ab initio ProteinStructure Prediction
Predicting a protein’s structure using only its amino acid
sequence is called ab initio structure prediction (ab
initio means “from the beginning” in Latin)
Biochemical research has developed scoring functions
called force fields that use the physicochemical
properties of amino acids introduced in the previous
lesson to compute the potential energy of a candidate
protein shape.
103.
Cont…
Problem: Findthe 3-D structure of protein having a
minimum E from a give sequence of A.A…
i.e., Needs Optimization
o An object maximizing or minimizing some function
subject to constraints
Ab Initio ProteinStructure Prediction
Protein structure prediction (PSP) is the prediction of the three-
dimensional structure of a protein from its amino acid sequence
i.e. the prediction of its tertiary structure from its primary
structure.
ab initio modelling conducts a conformational search under the
guidance of a designed energy function.
This procedure usually generates a number of possible
conformations (structure decoys), and final models are selected
from them.
106.
A successfulab initio modelling depends on three factors:
❖ An accurate energy function with which the native structure of a
protein corresponds to the most thermodynamically stable state,
compared to all possible decoy structures
❖ An efficient search method which can quickly identify the low-
energy states through conformational search
❖ Selection of native-like models from a pool of decoy structures.
Ab Initio Protein Structure Prediction…
107.
A local searchalgorithm for ab
initio structure prediction
Local search: nearby search
In protein structure prediction local search algorithm
find a protein structure that does not have minimum
free energy but that does have the property that no
“nearby” structures have lower energy
Local minimum is the decoy in search space that has
a smaller value of the optimization function than
neighboring points
Global minimum is the lowest energy
decoy/structure in among all the set of structures.
108.
Fundamental ways toavoid local
minima
How could we improve our local search algorithm for structure
prediction to avoid winding up in a local minimum?
A no of ways but two are fundamental;
First run the algorithm multiple times with different starting
conformations because the algorithm’s choice of initial conformation
has a huge influence on the final conformation
Second, every time we reach a local minimum, we could allow
ourselves to change the structure with some probability, thus giving
our local search algorithm the chance to “bounce” out of a local
minimum
Once again, randomized algorithms help us solve problems!
109.
QUARK
QUARK’s algorithmapplies a combination
of multiple scoring functions to look for the lowest
energy conformation across all of these functions.
111.
Conformational Search Methods
Successful ab initio modelling of protein structures depends on the availability of
a powerful conformation search method which can efficiently find the global
minimum energy structure for a given energy function with complicated energy
landscape.
Types:
❖ Monte Carlo Simulations
❖ Molecular Dynamics
❖ Genetic Algorithm
❖ Mathematical Optimization
112.
Monte Carlo Simulations
Its core idea is to use random samples of
parameters or inputs to explore the
behavior of a complex system or process.
113.
Molecular Dynamics
MDsimulation solves Newton’s equations of motion at each
step of atom movement, which is probably the most faithful
method depicting atomistically what is occurring in proteins.
Advantage: The method is therefore most-often used for
the study of protein folding pathways
Disadvantage: The long simulation time is one of the major
issues of this method, since the incremental time scale is
usually in the order of femtoseconds (10 15 s) while the
fastest folding time of a small protein (less than 100
residues) is in the millisecond range in nature.
114.
Genetic Algorithm
Thegenetic algorithm is a method for solving problems
that is based on natural selection, the process that drives
biological evolution.
❖ The genetic algorithm repeatedly modifies a population
of individual solutions.
❖ At each step, the genetic algorithm selects individuals at
random from the current population to be parents and
uses them to produce the children for the next generation.
❖ Over successive generations, the population "evolves“
toward an optimal solution.
























































































![ SWISS-MODEL is a structural bioinformatics web-server dedicated to homology
modeling of 3D protein structures.
3D protein structures provide valuable insights into the molecular basis of
protein function, allowing an effective design of experiments, such as site-
directed mutagenesis, studies of disease-related mutations or the structure
based design of specific inhibitors.
Automated homology modeling systems;
ModPipe (http://www.salilab.org)
CPHmodels (http://www.cbs.dtu.dk/services/CPHmodels/)
3D-JIGSAW (http://www.bmm.icnet.uk/~3djigsaw/)
ESyPred3D (http://www.fundp.ac.be/urbm/bioinfo/esypred/)
SDSC1 (http://cl.sdsc.edu/hm.html)].](https://image.slidesharecdn.com/bioinformatics-exammaterials-250416050939-2c3a80f1/75/Bioinformatics-Exam_Materials-pdf-by-uos-92-2048.jpg)






































































