Bioinformatics: An EmergingBranch of Biological Science
● Interdisciplinary field combining biology, information
technology, biology, chemistry, mathematics, statistics, and
computer science.
● Mainly involved in analyzing biological data and developing
new software using biological tools.
● Defined by NCBI, NLM, and NIH as the analysis, collection,
classification, manipulation, recovery, storage, and
visualization of biological information using computation
technology.
● First coined in 1960 by Paulien Hogeweg and Ben Hesper,
defining it as the study of information processes in biotic
systems.
INTRODUCTION TO BIOINFORMATICS
5.
Understanding Bioinformatics
● BioinformaticsDefinition: Bioinformatics is the application of computational
tools to manage and interpret biological data, enabling a deeper understanding
of genetic and molecular processes.
● Scope of Bioinformatics: It encompasses the development of algorithms and
software to analyze biological data, facilitating insights into genomics,
proteomics, and other biological disciplines.
● Importance in Research: Bioinformatics is pivotal in decoding plant genomes,
evolutionary studies, and drug discovery, contributing to advancements in
various fields.
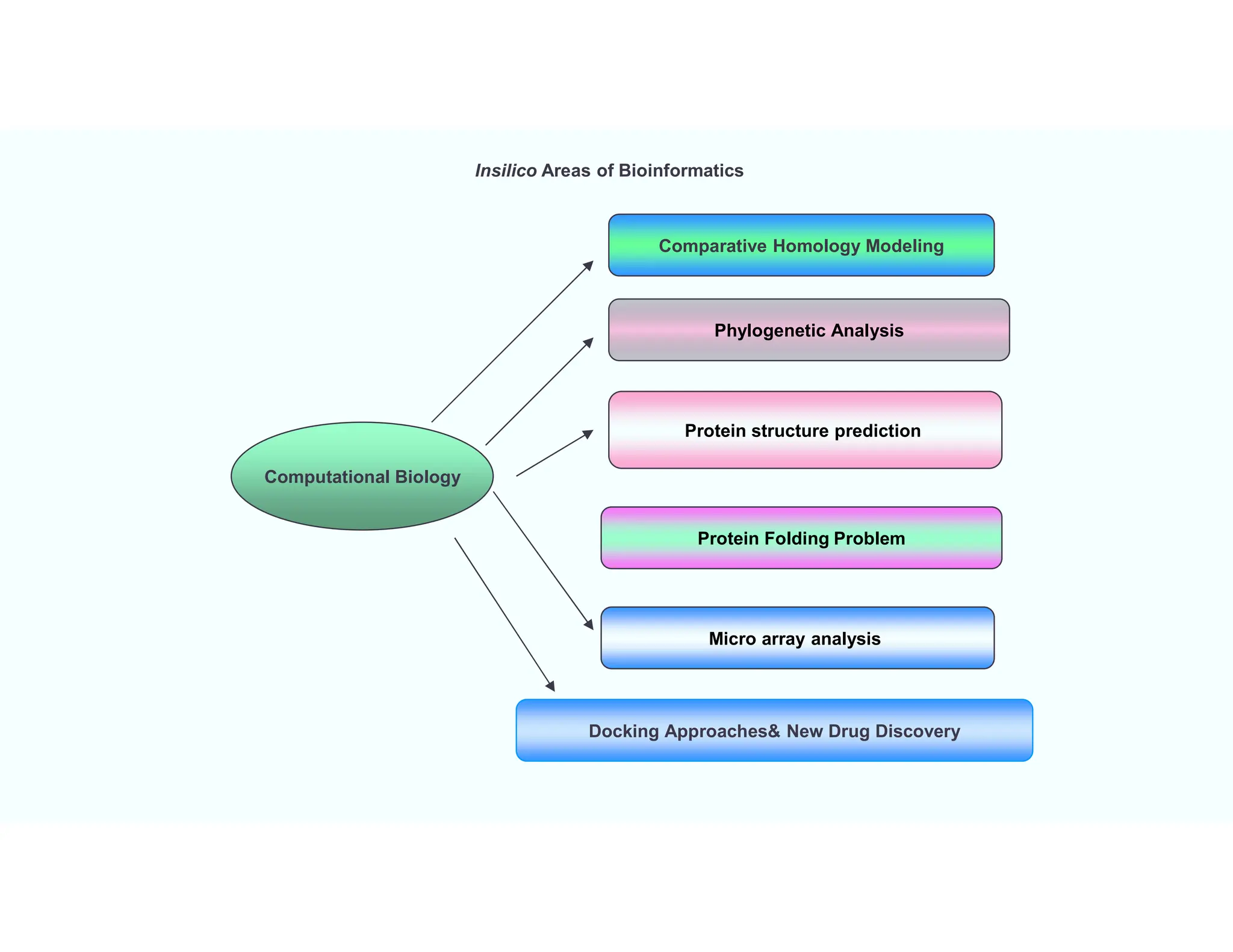
Insilico Areas ofBioinformatics
Computational Biology
Docking Approaches& New Drug Discovery
Protein structure prediction
Micro array analysis
Comparative Homology Modeling
Phylogenetic Analysis
Protein Folding Problem
8.
HISTORY AND SCOPEOF BIOINFORMATICS
• 1859 – The “On the Origin of Species”, published by Charles Darwin that introduced
theory of genetic evolution – allows adaptation over time to produce organisms best
suited to the environment.
• 1869 - The DNA from nuclei of white blood cells was first isolated by Friedrich Meischer.
• 1951 – Linus Pauling and Corey propose the structure for the alpha-helix and beta-
sheet.
• 1953 - Watson and Crick propose the double helix model for DNA based on x-ray data
obtained by Franklin and Wilkins.
• 1955 - The sequence of the first protein to be analyzed, bovine insulin, is announced by
F. Sanger.
• 1958 - The Advanced Research Projects Agency (ARPA) is formed in the US.
9.
• 1973 -The Brookhaven Protein Data Bank(PDB) is announced.
• 1987 - Perl (Practical Extraction Report Language) is released by Larry Wall.
•1988 - National Centre for Biotechnology Information (NCBI) founded at NIH/NLM.
•1990 - Human Genome Project launched BLAST program introduced by S. Karlin and S.F. Altshul.
• Tim Berners-Lee, a British scientist invented the World Wide Web in 1990.
•1992 - The Institute for Genome Research (TIGR), associated with plans to exploit sequencing
commercially through gene identification and drug discovery, was formed.
•2001 - The human genome (3,000 Mbp) is published.
•2010 :Completion of the 2010 Project: to understand the function of all genes within their cellular,
organism and evolutionary context of Arabidopsis thaliana.
HISTORY AND SCOPE OF BIOINFORMATICS
10.
Future Goals OfMolecular Biology and Bioinformatics Research
2050: To complete of the first computational model of a complete cell, or maybe
even already of a complete organism.
11.
Application of Bioinformatics
Genomics:
o Study of genomes through sequencing and analysis.
Proteomics:
o Study of the structure and function of proteins.
Transcriptomics:
o Analysis of RNA transcripts produced by the genome.
Metabolomics:
o Study of chemical processes involving metabolites.
12.
Bioinformatics Tools andTechniques
Sequence Alignment:
o Tools: BLAST, ClustalW
o Aligning DNA, RNA, or protein sequences to identify regions of similarity.
Genome Assembly:
o Tools: SPAdes, Velvet
o Piecing together short DNA sequences to reconstruct the original genome.
Structural Biology:
o Tools: PyMOL, Chimera
o Modeling and visualization of biomolecular structures.
13.
Genomics and Bioinformatics
Human Genome Project:
o Sequencing and mapping all genes in human DNA.
Applications:
o Disease gene identification, personalized medicine, evolutionary studies.
Proteomics and Bioinformatics
Protein Structure Prediction:
o Tools: SWISS-MODEL, Phyre2
o Predicting the 3D structure of proteins from amino acid sequences.
Protein-Protein Interactions:
o Tools: STRING, IntAct
o Understanding the networks and pathways in which proteins interact.
14.
Transcriptomics and Bioinformatics
RNA-Seq Analysis:
o Tools: HISAT2, DESeq2
o Analyzing gene expression levels across different conditions or time points.
Single-Cell RNA-Seq:
o Tools: Seurat, Scanpy
o Investigating gene expression at the single-cell level to understand cell heterogeneity.
Metabolomics and Bioinformatics
Metabolite Profiling:
o Tools: MetaboAnalyst, XCMS
o Identifying and quantifying metabolites in biological samples.
Pathway Analysis:
o Tools: KEGG, MetaCyc
o Mapping metabolic pathways and understanding metabolic changes in diseases.
.
15.
Bioinformatics in DrugDiscovery
Target Identification:
o Using bioinformatics to identify potential drug targets.
Virtual Screening:
o Tools: AutoDock, DOCK
o Screening large libraries of compounds to identify potential drug candidates.
Pharmacogenomics:
o Studying how genes affect a person’s response to drugs.
Bioinformatics in Agriculture
Crop Improvement:
o Genomic selection and breeding programs for better yield and disease resistance.
Microbial Genomics:
o Studying beneficial microbes in soil to enhance crop production.
Genetically Modified Organisms (GMOs):
o Bioinformatics in the development and assessment of GMOs.
.
16.
Internet and Bioinformatics
The Internet plays an important role in retrieving biological information.
Bioinformatics is an emerging new dimension of Biological science, including computer science , mathematics, and life
science.
The Computational part of bioinformatics used to optimize biological problems like (metabolic disorders, and genetic
disorders).
Computational part contains:
Computer Science
Operating System
Win 2000XPLinuxUnix
Database Development
Software & Tools Development
Software & Tools Application
17.
Internet and Bioinformatics
TheMathematical portion helps to understand the algorithms used in bioinformatics software and tools.
The mathematical portion used in Bioinformatics are :
Mathematics
Biostatistics
(HMM, ANN in secondary
structure prediction)
DiffrentiationIntigration
(Time and space complexity
E-value ,p-values in Blast)
Complex Mathematics Functions
(Fourier Transformation)
Matrices
(Sequence alignment, Blast Fast,
MSA & Phylogenetic Prediction)
What is Internet?
•The Internet is actually a network of networks, composed
of interconnected local and regional networks in over
100 countries using TCP/IP communication protocol.
• Transport Control Protocol/ Internet Protocol (TCP/IP)
• What is a protocol?
– A protocol is a set of rules defining communication
between systems
20.
What is Internet?
•Intranet: Use of TCP/IP to connect computers on an
organizational LAN
• Internet: Use of TCP/IP to interconnect such networks
• TCP/IP is the basic Internet protocol
21.
● Various applicationprotocols operate over TCP/IP
○ SMTP (Simple Mail Transfer Protocol)
○ HTTP(Hypertext Transfer Protocol)
○ IRC (Internet Relay Chat)
○ FTP (File transfer Protocol)
24.
INTERNET HISTORY
The trueorigins of the Internet lie with a
research project on networking at the
Advanced Research Projects Agency (ARPA)
of the US Department of Defense in 1969
named ARPANET.
The original ARPANET connected four
nodes on the West Coast, with the
immediate goal of being able to transmit
information on defense-related research
between laboratories.
25.
● In 1981,BITNET (‘‘Because It’s Time’’) was introduced, providing point-
to-point connections between universities for the transfer of
electronic mail and files.
● In 1982, ARPA introduced the Transmission Control Protocol (TCP)
and the Internet Protocol (IP);
● TCP/IP allowed different networks to be connected to and
communicate with one another.
● 1990s: World Wide Web (WWW) and the browser revolution.
● 2000s: Explosion of broadband, wireless, and mobile internet.
26.
TCP/IP
• Rules forinformation exchange between computers over
a network
• ‘Packet’ based – segment/ de-segment information
• Client-Server (Request/ Response)
– Web browser (client), Website (Server)
• TCP – Handles data part
• IP – Handles address part – Identification of every
computer on the Internet – IP address
27.
IP number ANDIP addresses
● IP number and IP addresses are unique, identifying one and only one
machine.
● The IP address is made up of four numbers separated by periods; for
example,
The IP address for the main file server at the National Center for
Biotechnology Information (NCBI) at the National Institutes of Health
(NIH) is 130.14.25.1.
The domain (130.14 for NIH),
The subnet (.25 for the National Library of Medicine at NIH), and
The machine itself (.1)
28.
How it works,and why it sometimes doesn't
● The Internet works by breaking up information into packets of data.
(TCP)
● Each packet of data is given an address and sent off on its merry
way.(IP)
● When the packets are received at the other end, they are reassembled
to give a faithful copy of the original data. (TCP)
29.
WEAKNESS AND STRENGTH
●Each packet gets passed by a machine to its neighbours which then decide to
pass it on, or pass it back.
● It's a strength because if a Internet node goes down (and this happens even
without nuclear strikes), the messages simply divert round the missing node.
This may mean taking a detour via a satellite link over the Indian Ocean, or
travelling via optical fibre via America. It really doesn't matter to you, the
Internet sorts it all out.
● It's a weakness because you usually need all your packets to reassemble the
original message, and if one takes a detour this may delay your whole message.
If one gets lost, it will usually prevent you getting the rest of the message.
30.
Domain name
● IPaddress are difficult to remember
● fully qualified domain name (FQDN) that is dynamically translated in the
background by domain name servers.
● Going back to the NCBI example, rather than use 130.14.25.1 to access the NCBI
computer, a user could instead use ncbi.nlm.nih.gov and achieve the same result.
Reading from left to right, notice that the IP address goes from least to most specific,
whereas the FQDN equivalent goes from most specific to least.
● The name of any given computer can then be thought of as taking the general form
computer. domain, with the top-level domain .
32.
CONNECTING TO THEINTERNET
● Copper Wires, Coaxial Cables, and Fiber Optics
33.
Content Providers vs.ISPs
● Once an appropriately fast and price-effective
connectivity solution is found, users will then need to
actually connect to some sort of service that will
enable them to traverse the Internet space.
● The two major categories in this respect are online
services and Internet service providers (ISPs).
34.
Content Providers
● Onlineservices, such as America Online (AOL),yahoo, msn and
CompuServe
● Once a connection has been made between the user’s computer and
the online service, one can access the special features, or content, of
these systems without ever leaving the online system’s host computer.
● Specialized content can range from access to online travel reservation
systems to encyclopedias that are constantly being updated—items that
are not available to nonsubscribers to the particular online service.
35.
ISP (Internet serviceproviders)
● Internet service providers take the opposite tack.
● Instead of focusing on providing content, the ISPs provide the
tools necessary for users to send and receive E-mail, upload
and download files, and navigate around the World Wide Web,
finding information at remote locations.
● E.g.. airtel,sify,BSNL, etc
36.
Internet Services
• E-Mail
•Telnet – remote login (e.g. library catalogue
access)
• FTP: File transfer (e.g. software packages)
• Web (HTTP): Hypertext linking/navigation
• IRC: Internet Relay Chat
• Internet telephony, mobile access, etc.
37.
ELECTRONIC MAIL
Its advantagesare many:
It is much quicker than the postal service or ‘‘snail mail.’’
Messages tend to be much clearer and more to the point than is
the case for
typical telephone or face-to-face conversations.
Recipients have more flexibility in deciding whether a response
needs to be sent immediately, relatively soon, or at all, giving
individuals more control over workflow.
It provides a convenient method by which messages can be filed or
stored.
There is little or no cost involved in sending an E-mail message.
FTP
● The FTP(File Transfer Protocol) utility program is
commonly used for copying files to and from other
computers.
41.
How to connect?
●To connect your local machine to the remote machine, type
ftp machinename
● where machinename is the full machine name of the remote
machine, e.g.,ftp.ncbi.nih.gov
Or
ftp machine number
● Where machine number is the ip number of the remote machine
e.g. 130.14.25.1
42.
Anonymous FTP
● Attimes you may wish to copy files from a remote machine on
which you do not have a login name. This can be done using
anonymous FTP.
● Login name: anonymous
● password: your own electronic mail address.
● This allows the remote site to keep records of the anonymous
FTP requests.
● you are only able to copy the files from the remote machine to
your own local machine; you are not able to write on the remote
machine or to delete any files there.
43.
Common FTP Commands
●? - to request help or information about the FTP commands
● ascii -To set the mode of file transfer to ASCII (this is the default and transmits seven bits per
character)
● binary -To set the mode of file transfer to binary (the binary mode transmits all eight bits per byte
and thus provides less chance of a transmission error and must be used to transmit files other
than ASCII files)
● bye - To exit the FTP environment (same as quit)
● cd- To change directory on the remote machine
● Close -To terminate a connection with another computer
● close brubeck - closes the current FTP connection with brubeck, but still leaves you within the FTP
environment.
● delete- to delete (remove) a file in the current remote directory (same as rm in UNIX)
● get -to copy one file from the remote machine to the local machine
● get ABC DEF- copies file ABC in the current remote directory to (or on top of) a file named DEF in
your current local directory.
● get ABC-copies file ABC in the current remote directory to (or on top of) a file with the same name,
ABC, in your current local directory.
44.
● help torequest a list of all available FTP commands
● lcd to change directory on your local machine (same as UNIX cd)
● ls to list the names of the files in the current remote directory
● mkdir to make a new directory within the current remote directory
● mget - to copy multiple files from the remote machine to the local machine; you are prompted for a y/n
answer before transferring each file
● mget * - copies all the files in the current remote directory to your current local directory, using the same
filenames. Notice the use of the wild card character, *.
● mput- to copy multiple files from the local machine to the remote machine; you are prompted for a y/n
answer before transferring each file
● open to open a connection with another computer
● open Brubeck - opens a new FTP connection with brubeck; you must enter a username and password for a
brubeck account (unless it is to be an anonymous connection).
● put to copy one file from the local machine to the remote machine
● pwd to find out the pathname of the current directory on the remote machine
● quit to exit the FTP environment (same as bye)
● rmdir to to remove (delete) a directory in the current remote directory
49.
World Wide Web
●In FTP we can download the files but we can’t view the file
content.
● This inherent drawback led to the development of a number of
distributed document delivery systems (DDDS),
● interactive client-server applications that allowed information
to be viewed without having to perform a download.
● The first generation of DDDS development led to programs like
Gopher, which allowed plain text to be viewed directly through a
client-server application
50.
● From thisevolved the most widely known and widely used DDDS
namely, the World Wide Web.
● it was conceived and developed at European
Nuclear Research Council (CERN) in 1989.
● That work led to a medium through which text, images, sounds,
and videos could be delivered to users on demand, anywhere in
the world.
51.
Navigation on theWorld Wide Web
● Advanced knowledge is not needed
● Website and webpage
● Clickable items ( hyperlink)
● Use of browers
● Advantage is view any site by entering the specific address
called as uniform resource locator
52.
Web Browsers
● Browserswere developed to use the
Hypertext Transport Protocol (http).
● A browser views a page written in HTML.
● They are the clients that communicate with
server using HTTP.
53.
Lynx- Academic computingservice at the university of Kansas which is
text mode runs on UNIX )
Mosaic-1993 at the National Center for supercomputing Applications
(NCSA)
● Google chrome Internet explorer and Firefox,
54.
● Cascading StyleSheets (CSS).
● JavaScript and other scripting languages
● Java, ActiveX and other interactive software
● plug-ins and add-ons. Plug-ins are commonly required
to handle special file types (e.g. audio and video files)
The National Centerfor
Biotechnology Information
Created in 1988 as a part of the
National Library of Medicine at NIH
– Establish public databases
– Research in computational biology
– Develop software tools for sequence analysis
– Disseminate biomedical information
Bethesda,MD
Goal of NCBI
(actof Congress, 1988)
● Create automated systems for knowledge about molecular
biology, biochemistry, and genetics
● Perform research into methods of analyzing molecular
biology data
● Enable researchers and medical care personnel to use the
systems developed
● Gather biotechnology information worldwide
60.
NCBI branches
● BasicResearch Branch- group of scientists who develop
algorithms and methods for analyzing molecular biology
data
● Information Resources Branch- maintains the infrastructure
at NCBI
● Information Engineering Branch- designs and builds
databases and software tools for molecular biology by
incorporating the new methods and approaches
61.
Databases and softwaretools designed by IEB
● GenBank, BLAST, PubMed, Entrez, LocusLink, GEO,
dbEST, dbSTS, Genome Resources, NCBI ToolBox,
Taxonomy Database, OMIM, dbSNP, dbMHC, Sequin,
BankIt, RefSeq Project, and many more
62.
Gathering biological data
●NCBI has to deal with gathering biological data,
which may come from different sources.
● So IEB developed the NCBI toolkit, which led to
creation of other softwares (GenBank, Entrez,
BLAST…).
● Those softwares can be used internally at NCBI to
process and analyze data that comes from variety
of sources.
● This allows NCBI to build and maintain the unified
databases.
63.
Accessibility of biologicaldata
● Softwares developed by IEB will be used by many
people.
● End-user scientists, bioinformatics specialists in
commercial, academic, or government settings, and
by academic researchers
● So it must be platform and format independent
● IEB chose to use ASN.1
64.
What is ASN.1?
●Abstract Syntax Notation number One
● The NCBI data model is often referred to as, and confused with, the
‘‘NCBI ASN.1’’ or ‘‘ASN.1 Data Model.’’
● Originated to make interaction from one computer to another
easier.
● Regardless of how data is represented, whatever the application,
whether complex or simple.
● As a matter of fact, NCBI uses ASN.1 to store and retrieve data such
as nucleotide and protein sequences, structures, genomes, and
MEDLINE records.
65.
Why they choseASN.1
● Molecular biology data comes from, and is used in variety
of environments.
● Data gathered and integrated by IEB will come from
many different sources, in many different models, and
they may change over time.
● Data should have longer life span than a particular
software tool or language.
● So IEB chose ASN.1, which is independent from hardware
or software architecture and language.
66.
Examples of theModel
● The GenBank flat file is a ‘‘DNA-centered’’ report, meaning that a region
of DNA coding for a protein is represented by a ‘‘CDS feature,’’ or
‘‘coding region,’’ on the DNA.
● A qualifier (/translation=“MLLYY”) describes a sequence of amino acids
produced by translating the CDS.
● mat_peptide, are occasionally used in GenBank flat files to describe
cleavage products of the (possibly unnamed) protein that is
described by a /translation
67.
Protein database
● Conversely,most protein sequence databases present a
‘‘protein-centered’’ view in which the connection to the
encoding gene may be completely lost or may be only
indirectly referenced by an accession number.
● Often times, these connections do not provide the exact
codon-to-amino acid correspondences that are important in
performing mutation analysis
68.
NCBI
● The NCBIdata model deals directly with the two
sequences involved: a DNA sequence and a protein
sequence.
● The translation process is represented as a link between
the two sequences rather than an annotation on one with
respect to the other.
● Protein-related annotations, such as peptide cleavage
products, are represented as features annotated directly
on the protein sequence.
69.
● Easy toanalyze the protein sequences .
● A collection of a DNA sequence and its translation products is
called a Nuc-prot set, and this is how such data is represented
by NCBI.
● The navigation provided by tools such as Entrez much more
directly reflects the underlying structure of such data. The
protein sequences derived from GenBank translations that are
returned by BLAST searches are, in fact, the protein sequences
from the Nuc-prot sets described above.
70.
● In thestandard GenBank format can also hide the
multiple sequence nature of some DNA sequences.
● For eg: For example, three genomic exons of a particular
gene are sequenced, and partial flanking, noncoding
regions around the exons may also be available, but the
full-length sequences of these intronic sequences may
not yet be available.
● Because the exons are not in their complete genomic
context, there would be three GenBank flatfiles in this
case, one for each exon.
71.
● The NCBIdata model defines a sequence type that directly
represents such a segmented series, called a ‘‘segmented
sequence.’’ Rather than containing the letters A, G, C, and T,
the segmented sequence contains instructions on how it
can be built from other sequences.
● The segmented sequence itself can have a name (e.g.,
HSDDT), an accession number, features, citations, and
comments, like any other GenBank record.
72.
● Data ofthis type are commonly stored in a so-called ‘‘Seg-set’’
containing the sequences HSDDT, HSDDT1, HSDDT2, HSDDT3 and all
of their connections and features.
● GenBank, EMBL, and DDBJ have recently agreed on a way to
represent these constructed assemblies, and they will be placed in a
new CON division, with CON standing for ‘‘contig’’ .
● In the Entrez graphical view of segmented sequences, the
segmented sequence is shown as a line connecting all of its
component sequences.
75.
The structure andcontent of the NCBI data
model
• There are two main reasons for putting data on a computer:
retrieval and discovery.
• NCBI uses four core data elements:
– bibliographic citations,
– DNA sequences,
– protein sequences, and
– Three-dimensional structures.
• In addition, two projects (taxonomy and genome maps) are also
included.
76.
PUBs: PUBLICATIONS ORPERISH
● It is the common process whereby scientific information is
reviewed, evaluated, distributed, and entered into the
permanent record of scientific progress.
● Publications serve as vital links between factual databases of
different structures or content domains (e.g., a record in a
sequence database and a record in a genetic database may
cite the same article).
77.
● Authors
● Articles
●Patents
● Citing Electronic Data submission
● MEDLINE (MUID) and PubMed
Identifiers(PMID)
○ PubMed Central
78.
● A Bioseqrepresents a single, continuous molecule of nucleic acid or
protein.
● It can be anything from a band on a gel to a complete chromosome.
● It can be a genetic or physical map.
● All Bioseqs have more common properties than differences.
● All Bioseqs must have at least one identifier, a Seq-id (i.e. Bioseqs
must be citable).
Biological Sequences
BIOSEQ
79.
Seq-id: Identifying theBioseq
● NCBI data model defines a whole class of object called sequence identifier.
● Every Bioseq MUST have at least one Seq-id, or sequence identifier.
80.
● This meansa Bioseq is always citable. You can refer
to it by a label of some sort.
● This is a crucial property for different software
tools or different scientists to be able to talk about
the same thing. There is a wide range of Seq-ids
and they are used in different ways.
● DDBJ/EMBL/GenBank share common accession no
● Where as PIR and SWISS-PORT have different
81.
Sequence identifiers
● GenBankgi|gi-number|gb|accession|locus
● EMBL Data Library gi|gi-number|emb|accession|locus
● DDBJ, DNA Database of Japan gi|ginumber|dbj|accession|locus
● NBRF PIR pir||entry
● Protein Research Foundation prf||name
● SWISS-PROT sp|accession|name
● Brookhaven Protein Data Bank (1) pdb|entry|chain
● Brookhaven Protein Data Bank (2)
entry:chain|PDBID|CHAIN|SEQUENCE
● Patents pat|country|number
● GenInfo Backbone Id bbs|number
● General database identifier gnl|database|identifier
● NCBI Reference Sequence ref|accession|locus
● Local Sequence identifier lcl|identifier
82.
Locus Name
● SCU49845
●Three characters(SCU)-
the organism
● Fourth and fifth
characters - other
group
designations(gene
product; segmented
entries),
● Last character -series
of sequential integers.
● Unique
● Instability
● LOCUS SCU49845 5028 bp DNA PLN 21-JUN-1999
● DEFINITION Saccharomyces cerevisiae TCP1-beta gene, partial cds,
and Axl2p
● (AXL2) and Rev7p (REV7) genes, complete cds.
● ACCESSION U49845
● VERSION U49845.1 GI:1293613
● KEYWORDS .
● SOURCE Saccharomyces cerevisiae (baker's yeast)
● ORGANISM Saccharomyces cerevisiae
● Eukaryota; Fungi; Ascomycota; Saccharomycotina;
Saccharomycetes;
83.
Accession Number
● uniqueidentifier for a sequence record
● No biological Meaning
● Stable
● Old: One Uppercase letter followed by 5 digits. (e.g., U12345)
● New: 2 Uppercase letter followed by 6 digits. (e.g., AF123456).
● Original accession number might become secondary to a newer accession number.
84.
Accession.Version
● Better seqeunce
identifier
●Combination of
accesion with version
number.
● "accession.version“
● e.g., U12345.1 → U12345.2
● when any change –
sequence receives a
new GI number AND an
increase to its version
number.
Accession Number onProtein
sequences
● Three letters followed by
five digits, a dot, and a
version number.
● Ex: AAA98665.1
● Any change to the
sequence data -the
version number will be
increased
● But the accession will
remain stable
● (e.g., AAA98665.1 will
change to AAA98665.2).
88.
Gi Number
● "GenInfoIdentifier“
● To all sequences processed into Entrez
● Integer number
● Given in addition to accession number
● If a sequence changes -a new GI number will be assigned.
● Separate GI number is also assigned to each protein translation within a nucleotide sequence record.
89.
○ nucleotide
sequence GI
numberis shown in
the VERSION field
of the database
record
○ protein sequence
GI number is
shown in the
CDS/db_xref field
of a nucleotide
database record,
and the VERSION
field of a protein
database record
90.
● The giis simply an integer number, sometimes referred
to as a GI number.
● It is an identifier for a particular sequence only.
● Suppose a sequence enters GenBank and is given an
accession number U00001. When the sequence is
processed internally at NCBI, it enters a database called
ID. ID determines that it has not seen U00001 before and
assigns it a gi number.eg 54
● update the record by changing the citation, so U00001
enters ID again. ID, recognizing the record, retrieves the
first U00001 and compares its sequence with the new
one. If the two are completely identical, ID reassigns gi 54
to the record. If the sequence differs it is given a new gi
number, say 88.
● At this time, ID marks the old record (gi 54) with the date
it was replaced and adds a ‘‘history’’ indicating that it was
replaced by gi 88.
● ID also adds a history to gi 88 indicating that it replaced
91.
● The ginumber serves three major purposes:
○ It provides a single identifier across sequences from many sources.
○ It provides an identifier that specifies an exact sequence. Eg. Anyone who
analyzes gi 54 and stores the analysis can be sure that it will be valid as long
as U00001 has gi 54 attached to it.
○ It is stable and retrievable.
(they can remap the former analysis)
92.
Reference Database
● RefSeqdatabase is a non-
redundant set of reference
standards
● That includes chromosomes,
complete genomic molecules
(organelle genomes, viruses,
plasmids), intermediate assembled
genomic contigs, curated genomic
regions, mRNAs, RNAs, and
proteins.
● RefSeq sequences are derived
from GenBank and provide non-
redundant curated data
● Updated as needed to maintain
current annotation or to
incorporate additional sequence
information.
93.
Reference Seq Id
●Accession.version,
prefixed
ex:NM_000001.1
● NT-constructed genomic contigs
NM_123456 -mRNAs NP_123456
-proteins NC_123456-
chromosomes
● BLAST OUTPUT:
● gi|4557284|ref|NM_000646.1|[4557284]
● gi -"GenBank Identifier
● 4557284 - gi number-
● ref -RefSeq -source database.
● NM_000646.1 -The RefSeq accession and version number.
94.
General Seq Id
●Genome Centres
● For identifying their sequences
● May never appear in the public
database
95.
Local Seq Id
●Used in data submission tool sequin
● During submission allocates number
● string or an integer
● local software to keep the local Seq-ids unique
● When completed submitted
mRNAs and Proteins
NM_123456Curated mRNA
NP_123456 Curated Protein
NR_123456 Curated non-coding RNA
XM_123456 Predicted mRNA
XP_123456 Predicted Protein
XR_123456 Predicted non-coding RNA
Gene Records
NG_123456 Reference Genomic Sequence
Chromosome
NC_123455 Microbial replicons, organelle , genomes,
human chromosomes
AC_123455 Alternate assemblies
Assemblies
NT_123456 Contig
NW_123456 WGS Supercontig
99.
Accession.Version Combined Identifier
●The International Nucleotide Sequence Database Collaboration (GenBank, EMBL, and DDBJ) introduced a
‘‘better’’ sequence identifier, one that combines an accession (which identifies a particular sequence record)
with a version number (which tracks changes to the sequence itself).
● Combining accession and version makes it clear to the casual user that a sequence has changed since an
analysis was done. Also, determining how many times a sequence has changed becomes trivial with a version
number. The
100.
Accession Numbers onProtein
Sequences
● The International Sequence Database Collaborators also started assigning accession. version numbers to
protein sequences within the records
● Protein accessions in these records consist of three uppercase letters followed by five digits and an integer
indicating the version.
101.
Reference Seq-id
● RefSeqrecords are a stable reference point for functional annotation, point mutation analysis, gene
expression studies, and polymorphism discovery.
102.
LOCUS U00089 816394bp DNA circular CON 10-MAY-
1999
DEFINITION Mycoplasma pneumoniae M129 complete genome.
ACCESSION U00089
VERSION U00089.1 GI:6626256
KEYWORDS .
SOURCE Mycoplasma pneumoniae.
ORGANISM Mycoplasma pneumoniae
Bacteria; Firmicutes; Bacillus/Clostridium group; Mollicutes;
Mycoplasmataceae; Mycoplasma.
REFERENCE 1 (bases 1 to 816394)
AUTHORS Himmelreich,R., Hilbert,H., Plagens,H., Pirkl,E., Li,B.C. and
Herrmann,R.
TITLE Complete sequence analysis of the genome of the bacterium Mycoplasma
pneumoniae
JOURNAL Nucleic Acids Res. 24 (22), 4420-4449 (1996)
MEDLINE 97105885
REFERENCE 2 (bases 1 to 816394)
AUTHORS Himmelreich,R., Hilbert,H. and Li,B.-C.
TITLE Direct Submission
JOURNAL Submitted (15-NOV-1996) Zentrun fuer Molekulare Biologie Heidelberg,
University Heidelberg, 69120 Heidelberg, Germany
FEATURES Location/Qualifiers
source 1..816394
/organism=“Mycoplasma pneumoniae”
/strain=“M129”
/db xref=“taxon:2104”
/note=“ATCC 29342”
CONTIG join(AE000001.1:1..9255,AE000002.1:59..16876,AE000003.1:59..10078,
AE000004.1:59..17393,AE000005.1:59..10859,AE000006.1:59..11441,…….
……………………………………………………………………………………………………………………
…………………………………………………………………………………………………………………..
103.
Classes of Bioseqs
•AVirtual Bioseq has the type of
molecule is known, but the sequence
is not known, and the precise length
may not be known (e.g., from the
size of a band on an electrophoresis
gel).
•A raw Bioseq contains a single
contiguous string of bases or
residues.
•A segmented Bioseq points to its
components, which are other raw or
virtual Bioseqs (e.g., sequenced
exons and undetermined introns).
104.
•A constructed sequencetakes its original
components and subsumes them, resulting in a
Bioseq that contains the string of bases or residues
and a ‘‘history’’ of how it was built.
•A map Bioseq places genes or physical markers,
rather than sequence, on its coordinates.
• A delta Bioseq can represent a segmented
sequence but without the requirement of assigning
identifiers to each component (including gaps),
although separate raw sequences can still be
referenced as components. The delta sequence is
used for unfinished high-throughput genome
sequences (HTGS) from genome centers and for
genomic contigs.
105.
BIOSEQ-SETs
COLLECTIONS OF SEQUENCES
●A biological sequence is often most appropriately stored in the context of other, related sequences.
For example, a nucleotide sequence and the sequences of the protein products it encodes naturally belong
in a set.
106.
● The mostcommon Bioseq-sets are
○ Nucleotide/Protein Sets:- containing a nucleotide and one or more protein
products, is the type of set most frequently produced by a Sequin data
submission.
○ Population and Phylogenetic Studies:- A major class of sequence
submissions represent the results of population or Phylogenetic studies.
○ Other Bioseq-sets: Seg set:- contains a segmented Bioseq and a Parts
Bioseq-set, which in turn contains the raw Bioseqs that are referenced by the
segmented Bioseq. This may constitute the nucleotide component of a Nuc-
prot set.
108.
SEQ-ANNOT:
ANNOTATING THE SEQUENCE
●Seq-annot, or sequence annotation, is a collection of information about a sequence, tied to specific regions
of Bioseqs through the use of Seq-loc's.
● A Bioseq can have many Seq-annot's associated with it.
● This allows knowledge from a variety of sources to be collected in a single place but still be attributed to the
original sources.
109.
● Multiple Seq-annotscan be placed on a Bioseq or on a Bioseq-set. Each Seq-annot
can have specific attribution.
● For example, PowerBLAST produces a Seq-annot containing sequence alignments,
and each Seq-annot is named based on the BLAST program.
110.
● Currently thereare three kinds of Seq-annot
○ Feature tables,
○ Alignments, and
○ Graphs
111.
Seq-feat: Features
● Asequence feature (Seq-feat) is a block of structured data explicitly attached to a region of a Bioseq
through one or two sequence locations (Seq-locs).
● The Seq-feat itself can carry information common to all features.
112.
● A featuremust always have a location.
● A coding region’s location usually starts at the ATG and
ends at the terminator codon.
● The location can have more than one interval if it is on a
genomic sequence and mRNA splicing occurs.
● For a coding region, the product Seqloc points to the
resulting protein sequence. This is the link that allows
the data model to separately maintain the nucleotide
and protein sequences, with annotation on each
sequence appropriate to that molecule.
● Features also have information unique to the kind of
feature. For example, the CDS feature has fields for the
genetic code and reading frame, whereas the tRNA
feature has information on the amino acid transferred.
113.
● Certain featuresdirectly model the central dogma of molecular biology and are
most likely to be used in making connections between records and in discovering
new information by computation.
● They are
○ GENES
○ RNA
○ CODING REGION
○ PROTEIN
114.
GENES
● The Genefeature indicates the location of a gene, a heritable region of
nucleic acid sequence that confers a measurable phenotype.
● That phenotype may be achieved by many components of the gene
being studied, including, but not limited to, coding regions, promoters,
enhancers, and terminators.
● The Gene feature is meant to approximately cover the region of nucleic
acid considered by workers in the field to be the gene. This admittedly
fuzzy concept has an appealing simplicity, and it fits in well with higher-
level views of genes such as genetic maps
115.
RNA
● An RNAfeature can describe both coding intermediates (e.g., mRNAs) and structural RNAs (e.g., tRNAs, rRNAs).
The locations of an mRNA and the corresponding coding region (CDS) completely determine the locations of
5 and 3 untranslated regions (UTRs), exons, and introns.
116.
Coding Regions
● ACoding Region (CDS) feature in the NCBI data model can be
thought of as ‘‘instructions to translate’’ a nucleic acid into its
protein product,via a genetic code .
● A coding region serves as a link between the nucleotide and
protein.
● The genetic code is assumed to be universal unless explicitly given
in the Coding Region feature. When the genetic code is not followed
at specific positions in the sequence—for example, when
alternative initiation codons are used in the first position, when
suppressor tRNAs bypass a terminator, or when selenocysteine is
added —the Coding Region feature allows these anomalies to be
indicated.
117.
Proteins
● A Proteinfeature names (or at least describes) a protein
or proteolytic product of a protein.
● A single protein Bioseq may have many Protein features
on it.
● It may have one over its full length describing a pro-
peptide, the primary product of translation. (The name in
this feature is used for the /product qualifier in the CDS
feature that produces the protein.) It may have a shorter
protein feature describing the mature peptide or, in the
case of viral polyproteins, several mature peptide
features.
● Signal peptides that guide a protein through a
membrane may also be indicated.
118.
Seq-graph: Graphs
● Graphsare the third kind of annotation that can go into Seq-annots.
● A Seq-graph defines some continuous set of values over a defined interval on a Bioseq.
● It can be used to show properties like G +C content, surface potential, hydrophobicity, or base accuracy over
the length of the sequence.
119.
● A Seq-descris meant to describe a Bioseq (or Bioseq-set) and place it in its biological and/or bibliographic
context.
● Seq-descrs apply to the whole Bioseq or to the whole of each Bioseq in the Bioseq-set to which the Seq-
descr is attached.
● Descriptors were introduced in the NCBI data model to reduce redundant information in records.
SEQ-DESCR:
DESCRIBING THE SEQUENCE
120.
BioSource: The BiologicalSource
• The BioSource includes information on the source organism (scientific name and common name)
• its lineage in the NCBI integrated taxonomy, and its nuclear and mitochondrial genetic code.
• It also includes information on the location of the sequence in the cell (e.g., nuclear genome or
mitochondrion)
• Additional modifiers (e.g., strain, clone, isolate, chromosomal map location).
121.
MolInfo: Molecule Information
•The MolInfo descriptor indicates the type of molecule [e.g., genomic, mRNA(usually isolated as cDNA), rRNA,
tRNA, or peptide],
• the technique with which it was sequenced (e.g., standard, EST, conceptual translation with partial peptide
sequencing for confirmation)
• the completeness of the sequence [e.g., complete, missing the left (5 or amino) end, missing both ends].
122.
FASTA Format
• FASTAformat contains a definition line and sequence characters and may be used as input to a variety of
analysis programs.
• The definition line starts with a right angle bracket (>) and is usually followed by the sequence identifiers in
a parsable form,
• example:
>gi|2352912|gb|AF012433.1|HSDDT2
• The remainder of the definition line, which is usually a title for the sequence, can be generated by software
from features and other information in a Nuc-prot set
123.
BLAST
• The BasicLocal Alignment Search Tool (BLAST; Altschul et al., 1990) is a popular method of ascertaining
sequence similarity.
• The BLAST program takes a query sequence supplied by the user and searches it against the entire database
of sequences maintained at NCBI.
• The output for each ‘‘hit’’ is a Seq-align, and these are combined into a Seq-annot.
124.
Entrez
● The Entrezsequence retrieval program (Schuler et al., 1996;) was designed to take advantage of connections
that are captured by the NCBI data model.
● The links in the data model allow retrieval of linked records.
125.
Sequin
• Sequin isa submission tool that takes raw sequence data and other biological information and assembles a
record for submission to one of the DDBJ/EMBL/GenBank databases.
• It makes full use of the NCBI data model and takes advantage of redundant information to validate entries.
• For example,
• Sequin can determine the coding region location (one or more intervals on the nucleotide that, through the
genetic code, produce the protein product).
• It compares the translation of the coding region to the supplied protein and reports any discrepancy.
• It also makes sure that each Bioseq has BioSource information applied to it
126.
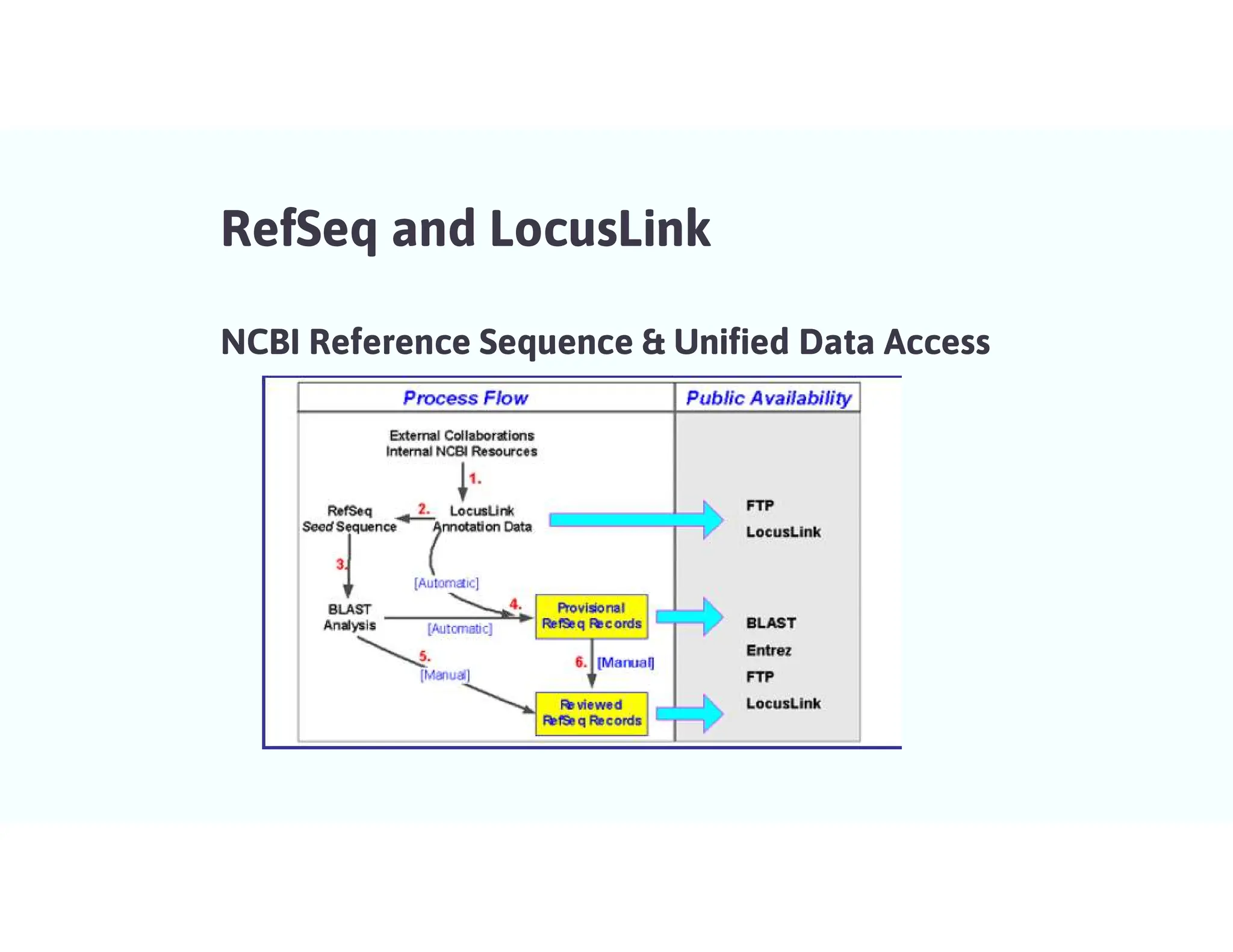
LocusLink
• LocusLink isan NCBI project to link information applicable to specific genetic loci from several disparate
databases.
• Information maintained by LocusLink includes official nomenclature, aliases, sequence
accessions,phenotypes, Enzyme Commission numbers, map information, and Mendelian Inheritance in Man
numbers.
• Each locus is assigned a unique identification number, which additional databases can then reference


























































































![Reference Seq Id
● Accession.version,
prefixed
ex:NM_000001.1
● NT-constructed genomic contigs
NM_123456 -mRNAs NP_123456
-proteins NC_123456-
chromosomes
● BLAST OUTPUT:
● gi|4557284|ref|NM_000646.1|[4557284]
● gi -"GenBank Identifier
● 4557284 - gi number-
● ref -RefSeq -source database.
● NM_000646.1 -The RefSeq accession and version number.](https://image.slidesharecdn.com/unit1compmolbio-250917234947-6d8df70d/75/Unit-1-computational-molecular-biology-for-engineers-93-2048.jpg)


























![MolInfo: Molecule Information
• The MolInfo descriptor indicates the type of molecule [e.g., genomic, mRNA(usually isolated as cDNA), rRNA,
tRNA, or peptide],
• the technique with which it was sequenced (e.g., standard, EST, conceptual translation with partial peptide
sequencing for confirmation)
• the completeness of the sequence [e.g., complete, missing the left (5 or amino) end, missing both ends].](https://image.slidesharecdn.com/unit1compmolbio-250917234947-6d8df70d/75/Unit-1-computational-molecular-biology-for-engineers-121-2048.jpg)





















































